{ "@context": "https://schema.org", "@graph": [ { "@id": "#issue", "@type": "PublicationIssue", "issueNumber": "1", "datePublished": "2024", "isPartOf": { "@id": "#periodical", "@type": [ "PublicationVolume", "Periodical" ], "name": "Cataloging & Classification Quarterly", "issn": [ "0004-4881", "2683-7226" ], "volumeNumber": "112", "publisher": "Revista de la Asociación Odontológica Argentina" } }, { "@type": "ScholarlyArticle", "isPartOf": "Revista de la Asociación Odontológica Argentina Volumen 112 Número 1", "mainEntityOfPage": "Asociación Odontológica Argentina", "datePublished": "2024-03-13", "headline": "Revista de la Asociación Odontológica Argentina", "image": "../img/raoa.jpg", "description": "", "sameAs": "https://doi.org/10.52979/raoa.1120432.1247", "about": [ "Works", "Catalog" ], "pageEnd": "", "pageStart": "", "name": "Enfermedad de Caffey después del primer año de vida. Caso clínico", "author":[ "Reynier Ramírez Suarez" , "Yuslaidy de los Ángeles López Consuegra" ] } ] }
Buscar
Enfermedad de Caffey después del primer año de vida. Caso clínico Caffey disease after the first year of life. Clinical case PATOLOGÍA | CASOS CLÍNICOS | OPEN ACCESS | PEER-REVIEWEDEnfermedad de Caffey después del primer año de vida. Caso clínicoCaffey disease after the first year of life. Clinical case
Autor/es: Reynier Ramírez Suarez, Yuslaidy de los Ángeles López Consuegra
Enfermedad de Caffey después del primer año de vida. Caso clínico
Caffey disease after the first year of life. Clinical case
Presentado: 11 de diciembre de 2023 Aceptado: 9 de enero de 2024 Publicado: 13 de marzo de 2024
Reynier Ramírez Suarez (a) Yuslaidy de los Ángeles López Consuegra (b)
a) Servicio de Cirugía Maxilofacial, Hospital Clínico Quirúrgico Docente Manuel Ascunce Domenech, Camagüey, Cuba b) Servicio de Cirugía Maxilofacial, Hospital Pediátrico Eduardo Agramonte Piña, Camagüey, Cuba
Resumen Objetivo: La enfermedad de Caffey o hiperostosis cortical infantil es una enfermedad rara que afecta uno o más huesos en los primeros meses de vida y debido a su baja incidencia está subdiagnosticada, y por tanto se aplican procedimientos invasivos innecesarios en su estudio y tratamiento. Se presenta un caso clínico atípico de enfermedad de Caffey en una paciente mayor de 1 año de edad y su resolución. Caso clínico: El servicio de Cirugía Maxilofacial del Hospital Provincial Pediátrico Eduardo Agramonte Piña de Camagüey, Cuba, atiende a una niña de 1 año y 10 meses que se encontraba hospitalizada por presentar una inflamación alarmante en la región facial y cervical precedida de un cuadro febril y dificultad para alimentarse. Se indicaron los estudios apropiados, cuyos resultados, junto a las características clínicas, permitieron diagnosticar la enfermedad de Caffey. Aunque sea una enfermedad rara, es importante estudiarla para realizar un correcto análisis de cada caso y diferenciarla de otras enfermedades que requieren de conductas terapéuticas agresivas. Palabras Claves: Cortical ósea, diagnóstico clínico, enfermedad de los huesos, enfermedad infantil, hiperostosis cortical infantil. Abstract Aim: Caffey’s disease or infantile cortical hyperostosis is a rare disease that affects one or more bones in the first months of life and due to its low incidence, it is underdiagnosed, and therefore unnecessary invasive procedures are applied in its study and treatment. An atypical clinical case of Caffey’s disease in a patient older than 1 year and its resolution is presented. Case report: The Maxillofacial Surgery service of the Eduardo Agramonte Piña Provincial Pediatric Hospital in Camagüey, Cuba, takes the case of a 1 year and 10-month-old female patient who was hospitalized for an alarming inflammation in the facial and cervical region, preceded by a fever and difficulty to eat. The appropriate studies were indicated, which results, together with the clinical characteristics, allowed the diagnosis of Caffey’s disease. Although it is a rare entity, it is important to study it to carry out a correct analysis of each case and differentiate it from other diseases that require aggressive therapeutic behaviors. Keywords: Bone disease, childhood disease, clinical diagnosis, cortical bone, infantile cortical hyperostosis. Introducción La enfermedad de Caffey o hiperostosis cortical infantil (HCI), fue reportada por primera vez por Roske en 1930 y descrita por Caffey y Silverman en 1945.(1,2) Es un trastorno óseo autolimitado caracterizado por la presencia de fiebre, irritabilidad, inflamación de los tejidos blandos y engrosamiento cortical de uno o más huesos.(3-5)
La HCI puede presentarse en su forma clásica infantil o en la forma prenatal. Esta última es la menos frecuente(2,6), aparece antes de las 35 semanas de edad gestacional y puede ser fatal debido a la prematuridad.
El curso natural de la enfermedad, el número de casos y la fisiopatología de la enfermedad están aún bajo investigación.(7) La variante infantil es frecuente en lactantes antes del quinto mes de vida y su período de resolución en los dos primeros años,(8) sin preferencia de sexo ni grupo étnico.1 Se desconoce su prevalencia exacta ya que se resuelve espontáneamente, elemento que favorece el subdiagnóstico de la enfermedad.(8) Su incidencia es de 48/100.000 y se considera fluctuante e influenciada por efectos ambientales. (8,9)
La etiología exacta de la HCI se desconoce en la actualidad, aunque existen varias teorías, entre las que se incluyen infecciones, alteraciones inmunológicas y anomalías genéticas.8 Se ha planteado su relación con la herencia autosómica dominante asociada a deficiencias en un tipo específico de gen (COLIAL, 17q21) que codifica la cadena alfa-1 del colágeno tipo I, y esto genera dudas sobre el posible vínculo de esta entidad a una colagenopatía, porque este es el gen responsable de patología como la osteogénesis imperfecta o el síndrome Ehlers-Danlos.(6,8,10)
La forma clásica o infantil leve rara vez ocurre después del primer año de edad,(2) se presenta generalmente con afectación asimétrica y la zona más recurrente es la mandíbula (70-90% de los casos), lo que puede ser empleado como criterio diagnóstico.(6)
La fase aguda tiene una duración aproximada de entre una y dos semanas y la mayoría de los pacientes presentan fiebre e irritabilidad, otras características clínicas son palidez, seudoparálisis dolorosa y pleuresía. (6,11)
En la mitad de los casos existe afectación clavicular, y en un 20-30% se produce la afectación de costillas y escápulas.(6) En una fase posterior se observan cambios radiológicos determinados por una reacción subperióstica con deformidades secundarias.(2,6,11) Las pruebas de laboratorio no son específicas para diagnosticar la enfermedad.(4) No se encuentran indicios serológicos de infecciones virales y bacterianas.(4,11) El estudio histopatológico define una reacción proliferativa del periostio de tipo inflamatorio, con lesiones inflamatorias de músculos y tejidos blandos vecinos. La piel y el tejido celular subcutáneo son normales.(11)
El diagnóstico de enfermedad de Caffey es fácil cuando la afectación es multifocal y asimétrica.(11) La enfermedad de Caffey es una enfermedad autolimitada y es posible que no necesite ninguna intervención,(8) sin embargo, los AINES podrían utilizarse en casos sintomáticos.(2,11,12) El tiempo de resolución puede variar entre semanas y años, y en este período puede modificarse la zona inflamatoria. Se registraron algunos casos raros de recurrencia en la adolescencia y la edad adulta.2
La identificación oportuna de esta rara pero importante enfermedad puede prevenir retrasos en el diagnóstico o el uso prolongado de antibióticos.(3)
El objetivo de este artículo es presentar las características clínicas-radiográficas de un caso atípico de la enfermedad de Caffey.
Caso clínico Se solicitó una interconsulta al servicio de Cirugía Maxilofacial del Hospital Provincial Pediátrico Eduardo Agramonte Piña de Camagüey, Cuba, por el servicio de Pediatría para evaluar una paciente de 1 año y 10 meses de edad y sexo femenino que cumplía su segundo día de estancia hospitalaria por presentar fiebre con valores superiores a 38 grados, irritabilidad y dificultad para alimentarse. El médico a cargo informó sobre la administración de antipiréticos orales y la aparición de una inflamación en la región cérvico-facial izquierda en las últimas 24 horas.
Los autores contaron con el consentimiento informado por escrito de los padres para realizar los procedimientos diagnósticos y terapéuticos necesarios como así también para la publicación del caso y el uso de fotografías.
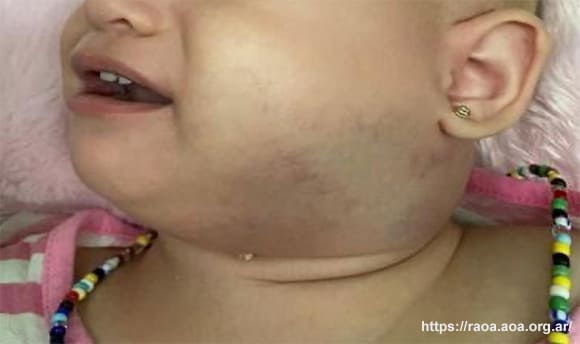
Al examen físico se constató tumefacción indurada en la región submandibular y retromandibular izquierda con extensión cervical y la zona afectada se encontró con alteraciones en la coloración (fig. 1).
Figura 1. Tumefacción en región submandibular y retromandibular izquierda con extensión cervical. La zona se muestra con cambios en la coloración. La publicación de la imagen fue autorizada por los padres.
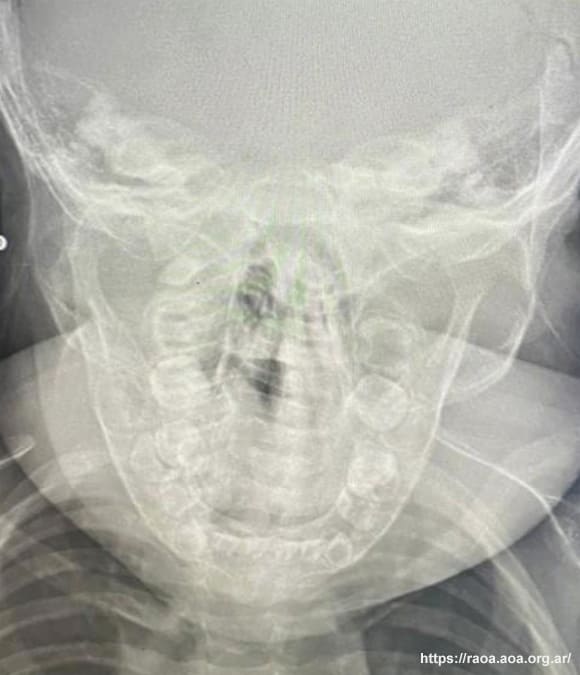
El estudio radiológico de la proyección anteroposterior de cráneo mostró engrosamiento cortical a nivel del ángulo y rama ascendente mandibular izquierda (fig. 2). El estudio de laboratorio resultante registró leucocitosis leve y aumento de los niveles de fosfatasa alcalina y proteína C reactiva. El hematocrito se halló en valores inferiores al rango normal.
Figura 2. Vista anteroposterior de cráneo: se observa engrosamiento cortical en ángulo mandibular y rama ascendente (lado izquierdo).
Una vez planteada la impresión diagnóstica de enfermedad de Caffey se decide mantener con ibuprofeno 5 ml cada 8 horas vía oral. La paciente egresa del centro 48 horas después, al constatar una mejoría notoria del estado general. Se continuó la evaluación en consulta externa y a las seis semanas se encontró remisión del proceso inflamatorio en casi su totalidad (fig. 3). El aspecto normal de la región facial se recuperó a los 4 meses.
Figura 3. Imagen clínica de la paciente a las 4 semanas de iniciados los síntomas que muestra la regresión parcial del proceso inflamatorio. La publicación de la imagen fue autorizada por los padres.
Discusión La enfermedad de Caffey puede presentarse en dos formas: una forma infantil leve (clásica) y la forma prenatal grave.(8) Esta última se considera una variante poco común y es probable que sea una patología completamente separada.(8)
La forma clásica es un proceso autolimitado, casi exclusivo de lactantes, aunque se ha descrito en niños mayores. Puede seguir un curso recidivante que finaliza antes de los tres años.(9) El comienzo tiene un promedio de edad que se sitúa entre el mes y los dos meses de vida, aunque se han descrito casos neonatales y otros más tardíos, pero casi siempre de inicio anterior a los cinco o seis meses de edad.(8,9) El caso de la paciente presentado en el trabajo superó el año de edad.
El desarrollo de la HCI puede ser esporádico o transmitirse por herencia autosómica dominante mediante penetrancia incompleta.(6,9) Los casos de afectación familiar se asocian con una mutación sin sentido del gen COL1A1 (3040C a T) en el cromosoma 17q21 que da como resultado una sustitución de arginina por cisteína (R836C) en la triple hélice de la cadena alfa-1 del colágeno tipo 1.(2,6) El colágeno tipo 1 es fundamental en la mineralización ósea y representa aproximadamente la décima parte de la matriz ósea, proporciona fuerza e interactúa con las proteínas para formar la matriz extracelular necesaria para el funcionamiento celular.(2) Se teoriza en la patogenia un fenómeno de hipoxia, necrosis local y reacción subperióstica sin precisar con exactitud los desencadenantes.(9) El daño hipóxico del periostio, resultante de defectos hereditarios de las arteriolas, conlleva a la proliferación de células subperiósticas y fibrosis de la médula ósea que origina un proceso de hematopoyesis extramedular hepática e hipertensión portal.(13)
La enfermedad de Caffey suele presentar una tríada de signos, síntomas e imagen radiográfica.(8)
La forma de presentación más habitual se caracteriza por inflamación dolorosa en el tejido blando suprayacente al hueso afectado que suele preceder al engrosamiento cortical, irritabilidad, fiebre sin claro foco, dificultad para alimentarse y dolor en lactantes.(2,6,8) Según Kirby,2 estas inflamaciones se vuelven duras y fijadas al hueso y también pueden ser rojas y dolorosas, en contraposición a lo planteado por Refai et al.(8) que aseveran que la hinchazón se presenta sin enrojecimiento ni calor. En el caso clínico del presente trabajo pudo observarse la región afectada enrojecida.
En ese sentido se puede acordar con Tilva et al.(14) cuando plantean que la HCI puede presentarse con signos clínicos inconsistentes que se asemejan a los de la osteomielitis, por lo que es fundamental tener un alto grado de sospecha.
El sitio afectado más común según su orden es la mandíbula, escápula, clavícula y los huesos largos.(2,8)
Existen reportes de parálisis del nervio facial y en caso de dolor intenso puede presentarse pseudoparálisis y hallazgos clínicos raros, como obstrucción nasal, proptosis y disfagia.(8)
Si bien la radiografía no es concluyente para la enfermedad, se considera el estudio más valioso para excluir los diagnósticos diferenciales importantes y debe ser la modalidad principal de investigación y seguimiento.(4,8) Un rasgo característico es el engrosamiento cortical gradual y la nueva formación de hueso cortical debajo de las regiones de inflamación de los tejidos blandos que están afectados.(2,4,8) La resonancia magnética es más específica sobre las reacciones de los tejidos blandos y el edema, y permite excluir procesos sépticos y tumores.(8) Los cambios tardan entre 15 y 20 días en aparecer en las radiografías, lo que dificulta el diagnóstico de la HCI en las primeras etapas.(2)
No existe una prueba de laboratorio específica para diagnosticar la HCI, pero se avala el componente inflamatorio al presentarse con leucocitosis, aumento de la velocidad de sedimentación globular (ESR) y fosfatasa alcalina (ALP).(8) La histología de la HCI evoluciona según la fase de la enfermedad, desde alteraciones limitadas al periostio hasta la formación de hueso nuevo.(2)
Se realiza el diagnóstico diferencial para descartar neoplasias malignas, hipervitaminosis A, hipoparatiroidismo, parotiditis, osteomielitis, administración de prostaglandinas, sarcoma de Ewing, escorbuto u otros defectos en la síntesis de colágeno.(2,8) Lee et al.(12) reportan un caso de un niño de 8 meses con reacción perióstica extensa en huesos largos por abuso infantil que fue considerado inicialmente como HCI.
El diagnóstico de la paciente del presente estudio se basó en la presencia de fiebre e irritabilidad, la instauración del edema, las características radiográficas y las pruebas de laboratorio alteradas. La edad de la paciente no contribuyó en gran medida a orientar el diagnóstico, ya que según Kirby et al.(12) es rara esta enfermedad después del primer año. De igual forma la presencia de enrojecimiento en la región afectada no es frecuente según la literatura.
No existe ningún tratamiento específico contra la enfermedad.(7) Se pueden indicar antiinflamatorios no esteroideos o corticoterapia para garantizar el alivio del dolor.(7,11) En el trabajo de Campañá et al.(7) se presentan tres casos que fueron tratados con cefalosporinas de tercera generación sin cambios en el curso clínico ni humoral. En el caso de esta paciente no se emplearon antimicrobianos y su evolución fue favorable.
El conocimiento de la existencia de esta rara afección y su perfil clínico-radiológico típico protegerá al paciente de ser sometido a múltiples investigaciones y tratamientos invasivos innecesarios.(15)
Declaración de conflictos de intereses: Los autores declaran no tener conflicto de intereses en relación con este artículo. científico.
Fuentes de financiamiento: Este estudio fue financiado exclusivamente por los autores.
Caffey J. Infantile cortical hyperostosis; a review of the clinical and radiographic features. Proc R Soc Med 1957;50:347-54.
Kirby K, Ponnarasu S, Alsaleem M, Wright JE. “Infantile cortical hiperostosis”, en StatPearls, Treasure Island (FL), StatPearls Publishing, 2024[citado el 12 de septiembre de 2023]. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK532878/
Merdler-Rabinowicz R, Grinberg A, Jacobson JM, Somekh I, Klein C, Lev A, et al. Fetuin-A deficiency is associated with infantile cortical hiperostosis (Caffey disease). Pediatr Res 2019;86:603-7. https://doi.org/10.1038/s41390-019-0499-0
Kamoun-Goldrat A, le Merrer M. Infantile cortical hyperostosis (Caffey disease): a review. J Oral Maxillofac Surg 2008;66:2145-50. https://doi.org/10.1016/j.joms.2007.09.007
Campañá Cobas NG, Santana Garay J, Duran Álvarez S, Vázquez Ríos B, Moreno Díaz H. Hiperostosis cortical infantil. A propósito de tres casos. Rev Cubana Pediatr 2006;78:2.
Refai AH, Taha WS, Almahdi H, Arabi H. Caffey’s disease (infantile cortical hiperostosis. Case report, MRI findings, and review of the literature. J Musculoskelet Surg Res 2018;2:173-6. https://doi.org/10.4103/jmsr.jmsr_42_18
Ludman A, Bravo M, Moguillansky S. Descripción del caso presentado en el número anterior: Hiperostosis cortical infantil: Enfermedad de Caffey. Arch Argent Pediatr 2010[citado el 11 de septiembre de 2023];108:360-2. Disponible en: https://www.sap.org.ar/docs/publicaciones/archivosarg/2010/v108n4a16.pdf
Lee DY, Kim WJ, Kim B, Nho JH, Hong CH, Lee SM, et al. Differential diagnosis between child abuse and infantile cortical hyperostosis: A case report and literature review. Int J Environ Res Public Health 2021;18:12269. https://doi.org/10.3390/ijerph182212269
Kim ST, Kim H, Kim HH, Lee NH, Han Y, Sung SI, et al. A rare case of lethal prenatal-onset infantile cortical hyperostosis. Yonsei Med J 2019;60:484-6. https://doi.org/10.3349/ymj.2019.60.5.484
Tilva H, Kanjiya A, Umate R, Wanjari MB, Tivaskar S. A rare case of infantile corticale hyperostosis (ICH) of the bilateral tibia or Caffey disease. Cureus 2023;11:e33655. https://doi.org/10.7759/cureus.33655
Cortical ósea diagnóstico clínico enfermedad de los huesos enfermedad infantil hiperostosis cortical infantil Bone disease childhood disease clinical diagnosis cortical bone infantile cortical hyperostosis
Citar este artículoRamírez Suarez R, López Consuegra Y. Enfermedad de Caffey después del primer año de vida. Caso clínico. Rev Asoc Odontol Argent. 2024-Mar-13;112(1):e1120432. https://doi.org/10.52979/raoa.1120432.1247 Copiar Cita
PDF | Subir | Home
X Twitter | WhatsApp | Facebook | E-mail
Las obras publicadas en este sitio están bajo una Licencia Creative Commons Atribución-NoComercial 4.0 Internacional
Revista indexada en:
Los artículos científicos de RAOA se identifican con DOI (Digital Object Identifier) Crossref.
Visitas al artículo: 1.062
Lecturas en línea: 884
Descargas del artículo: 178
Accesos por DOI: 201
Database ID: 2024005008
Cortical ósea diagnóstico clínico enfermedad de los huesos enfermedad infantil hiperostosis cortical infantil Bone disease childhood disease clinical diagnosis cortical bone infantile cortical hyperostosis
Auspiciantes del Website:
Revista de la Asociación Odontológica Argentina
La Revista de la Asociación Odontológica Argentina, RAOA, es la principal vía de expresión de la producción científica argentina en odontología y un vínculo de unión y medio de información para los colegas e instituciones del país y del exterior. Versión impresa ISSN 0004-4881 Versión electrónica ISSN 2683-7226. DOI: 10.52979/raoa.1898 Título abreviado: Rev Asoc Odontol Argent